Yi Zhang Ph.D.

Address: 450 Brookline Ave,
Boston 02215 USA
Email: yiz [AT] ds.dfci.harvard.edu
[ Google Scholar ] [ Harvard Profile ]
 https://orcid.org/0000-0002-7453-6188
https://orcid.org/0000-0002-7453-6188I am a Research Fellow at the Department of Data Science, Dana-Farber Cancer Institute and Harvard University T.H. Chan School of Public Health. I joined Dr. X. Shirley Liu's lab in July 2019 and also co-mentored by Dr. Myles Brown and Dr. Xihong Lin. I did my PhD in Bioengineering in Dr. Jun S. Song's group at University of Illinois at Urbana-Champaign working for NIH Big Data to Knowledge (BD2K) U54 center.
I have developed computational biology and statistical learning methods to decipher human genetic variants associated with cancer risk. I have also been developing machine learning methods for single-cell genomics data to understand cells in tumor.
I also enjoy working on patient genomics data to understand disease and identify therapeutic targets. Our collaborators include Dr. Catherine J. Wu at DFCI and Dr. David E. Fisher at MGH.
My research interests are to develop statistical and machine learning methods for data-driven discoveries in biomedicine.
For fun, I enjoy hiking, music and wildlife photography.
I'll join Duke University in Jan 2024 as an Assistant Professor in Department of Neurosurgery, and Department of Bioinformatics & Biostatistics.
Grad students and postdocs in the areas of computational biology, bioinformatics, cancer genomics, and machine learning - Welcome to join us!
Website: Computational Biology Lab @ Duke
Education
Ph.D. Bioengineering, Aug. 2014 - May 2019
- Department of Bioengineering, University of Illinois at Urbana-Champaign
- Committee: Dr. Jun S. Song, Dr. Saurabh Sinha, Dr. Dave S. Zhao, Dr. Pablo Perez-Pinera
Bachelor of Science, Biosciences, Sep. 2010 - Jul. 2014
- Life Sciences Talent Program, University of Science and Technology of China
- Department of Life Sciences, University of Science and Technology of China
Publications
- Zhang Y, Xiang G, Fan J, Wang C, Lynch A, Zeng Z, Zhang W, Kang J, Jiang A, Gu S, Wan C, Zhang B, Brown M#, Liu XS#, Meyer C#. "MetaTiME: Meta-components of the Tumor Microenvironment." Nature Communications (2022) Accepted in pinciple. Preprint on bioRxiv. Tweetorial on twitter.
- Choi Y*, Erlich T*, Franque M, Rachmin I, Flesher J, Schiferle E, Zhang Y, Silva M, Jiang A, Dobry A, Lacher S, Freund O, Feder E, Cortez J, Ryu S, Samuels Y, Zakka L, Azin M, Burd C, Shapless NN, Liu XS, Meyer C, Austen W, Bojovic B, Certrulo C, Mihm M, Hoon D, Demehri S, Hawryluk E, Fisher DE#. “Topical therapy for regression and melanoma prevention of congenital giant nevi.” Cell (2022) Accepted .
- Qiu X*, Boufaied N*, Hallal T, Feit A, Polo A de, Luoma AM, Alahmadi W, Larocque J, Zadra G, Xie Y, Gu S, Tang Q, Zhang Y, Syamala S, Seo JH, Bell C, O’Connor E, Liu Y, Schaeffer E, Karnes R, Weinmann S, Davicioni E, Morrissey C, Cejas P, Ellis L, Loda M, Wucherpfennig K, Pomerantz M, Spratt D, Corey E, Freedman M, Liu XS, Brown M, Long H#, Labbé D#. “MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets.” Nature Communications. (2022) Accepted.
- Zeng Z#, Wong C, Yang L, Ouardaoui N, Li D, Zhang W, Gu S, Zhang Y, Liu Y, Wang X, Fu J, Zhou L, Zhang B, Kim S, Yates K, Brown M, Freeman G, Uppaluri R, Manguso R, Liu XS#. “TISMO: syngeneic mouse tumor database to model tumor immunity and immunotherapy response.” Nucleic Acids Research. (2022)
- Zhou L*, Zeng Z*, Egloff AM, Zhang F, Guo F, Campbell KM, Du P, Fu J, Zolkind P, Ma X, Zhang Z, Zhang Y, Wang X, Gu S, Riley R, Nakahori Y, Keegan J, Haddad R, Schoenfeld J, Griffith O, Manguso RT, Lederer JA, Liu XS#, Uppaluri R#. “Checkpoint blockade-induced CD8+ T cell differentiation in head and neck cancer responders.” Journal for ImmunoTherapy of Cancer. (2022)
- Penter L, Zhang Y, Savell A, Huang T, Cieri N, Thrash EM, Kim-Schulze S, Jhaveri A, Fu J, Ranasinghe S, Li S, Zhang W, Hathaway E, Nazzaro M, Kim H, Chen H, Thurin M, Rodig S, Severgnini M, Cibulskis C, Gabriel S, Livak K, Cutler C, Antin J, Nikiforow S, Koreth J, Ho V, SArmand P, Ritz J, Steicher H, Neuberg D, Hodi F, Gnjatic S, Soiffer R, Liu SX, David M, Bachireddy P, Wu CJ#. “Molecular and cellular features of CTLA-4 blockade for relapsed myeloid malignancies after transplantation.” Blood. (2021)
- Zhang Y, Liu Y, Liu XS#. “Neural network architecture search with AMBER.” Nature Machine Intelligence. News & Views. (2021) 3:372–3.
- Gu SS*, Zhang W*, Wang X*, Jiang P*, Traugh N, Li Z, Meyer C, Stewig B, Xie Y, Bu X, Manos M, Front-Tello A, Gjini E, Lako A, Lim K, Conway J, Tewari A, Zeng Z, Sahu A, Tokleim C, Weirather J, Fu J, Zhang Y, Kroger B, Liang J, Cejas P, Freeman G, Rodig S, Long H, Gewurz B, Hodi F, Brown M#, Liu XS#. “Therapeutically Increasing MHC-I Expression Potentiates Immune Checkpoint Blockade.” Cancer Discovery (2021) 11:1524–41.
- Manjunath M*, Yan J*, Youn Y, Drucker KL, Kollmeyer TM, McKinney AM, Zazubovich V, Zhang Y, Costello J, Eckel-Passow J, Selvin P, Jenkins R, Song JS#. “Functional analysis of low-grade glioma genetic variants predicts key target genes and transcription factors.” Neuro-Oncology. (2021) 23:638–49.
- Baur B, Schreiber J, Shin J, Zhang S, Zhang Y, Manjunath M, Song JS, Noble WS, Roy S#. “ Leveraging epigenomes and three-dimensional genome organization for interpreting regulatory variation. ” Under Review. bioRxiv (2021).08.29.458098.
- Gu SS, Wang X, Hu X, Jiang P, Li Z, Traugh N, Bu X, Tang Q, Wang C, Zeng Z, Fu J, Meyer C, Zhang Y, Cejas P, Lim K, Wang J, Zhang W, Tokheim C, Sahu A, Xing X, Kroger B, Ouyang Z, Long H, Freeman G, Brown M, Liu XS. “Clonal tracing reveals diverse patterns of response to immune checkpoint blockade.” Genome Biology. (2020)
- Manjunath M, Zhang Y, Zhang S, Roy S, Perez-Pinera P, Song JS. “ABC-GWAS: Functional Annotation of Estrogen Receptor-Positive Breast Cancer Genetic Variants.” Frontiers in Genetics. (2020);0:730.
- Zhang Y, Manjunath M, Yan J, Baur BA, Zhang S, Roy S, et al. “The cancer-associated genetic variant rs3903072 modulates immune cells in the tumor microenvironment.” Frontiers in Genetics (2019);
- Zhang Y. “Functional interpretation of cancer-associated genetic variants.” University of Illinois at Urbana-Champaign. (2019) (Dissertation)
- Zhang Y, M. Manjunath*, S. Zhang, D. Chasman, S. Roy, and J.S. Song. "Integrative genomic analysis predicts causative cis-regulatory mechanisms of the breast cancer-associated genetic variant rs4415084." Cancer Research, 78(7), 1579-1591, (2018).
-
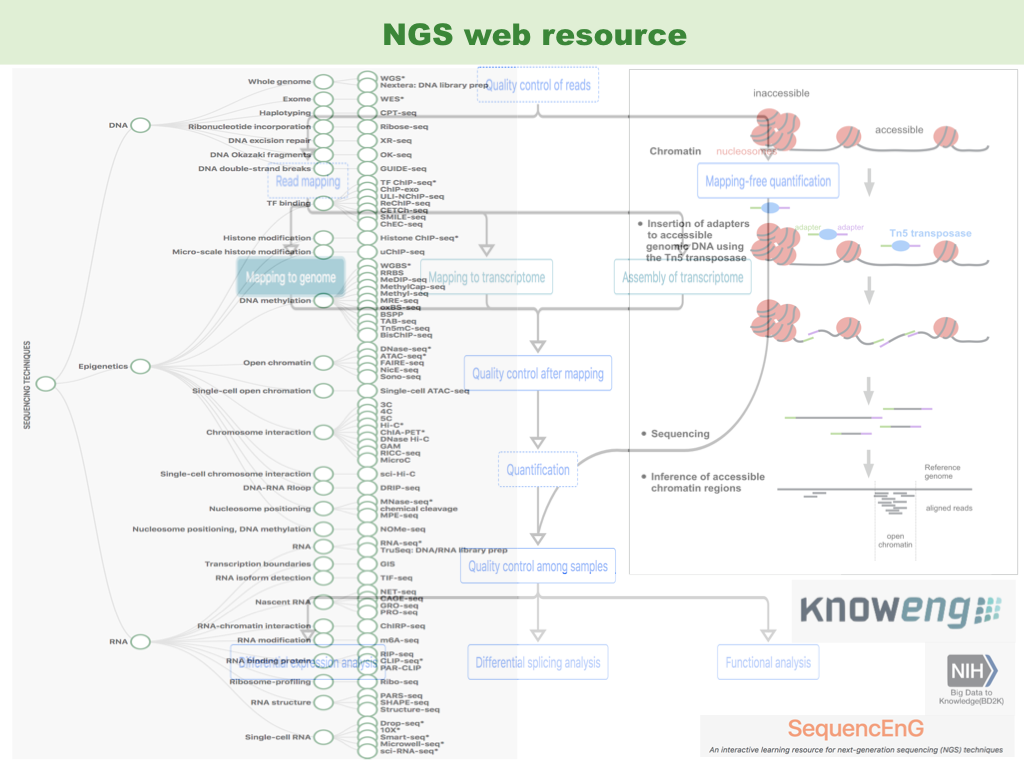
Zhang Y, Manjunath M, Kim Y, Heintz J, Song JS. "SequencEnG: an Interactive Knowledge Base of Sequencing Techniques." Bioinformatics, bty794, (2019).
[ SequencEnG page ] - Manjunath M, Zhang Y, Kim Y, Yeo SH, Sobh O, Russell N, et al. "ClusterEnG: an interactive educational web resource for clustering and visualizing high-dimensional data." PeerJ Computer Science 4, e155, (2018). [ ClusterEnG page ] (*co-first authors, #corresponding author )
Selected Projects
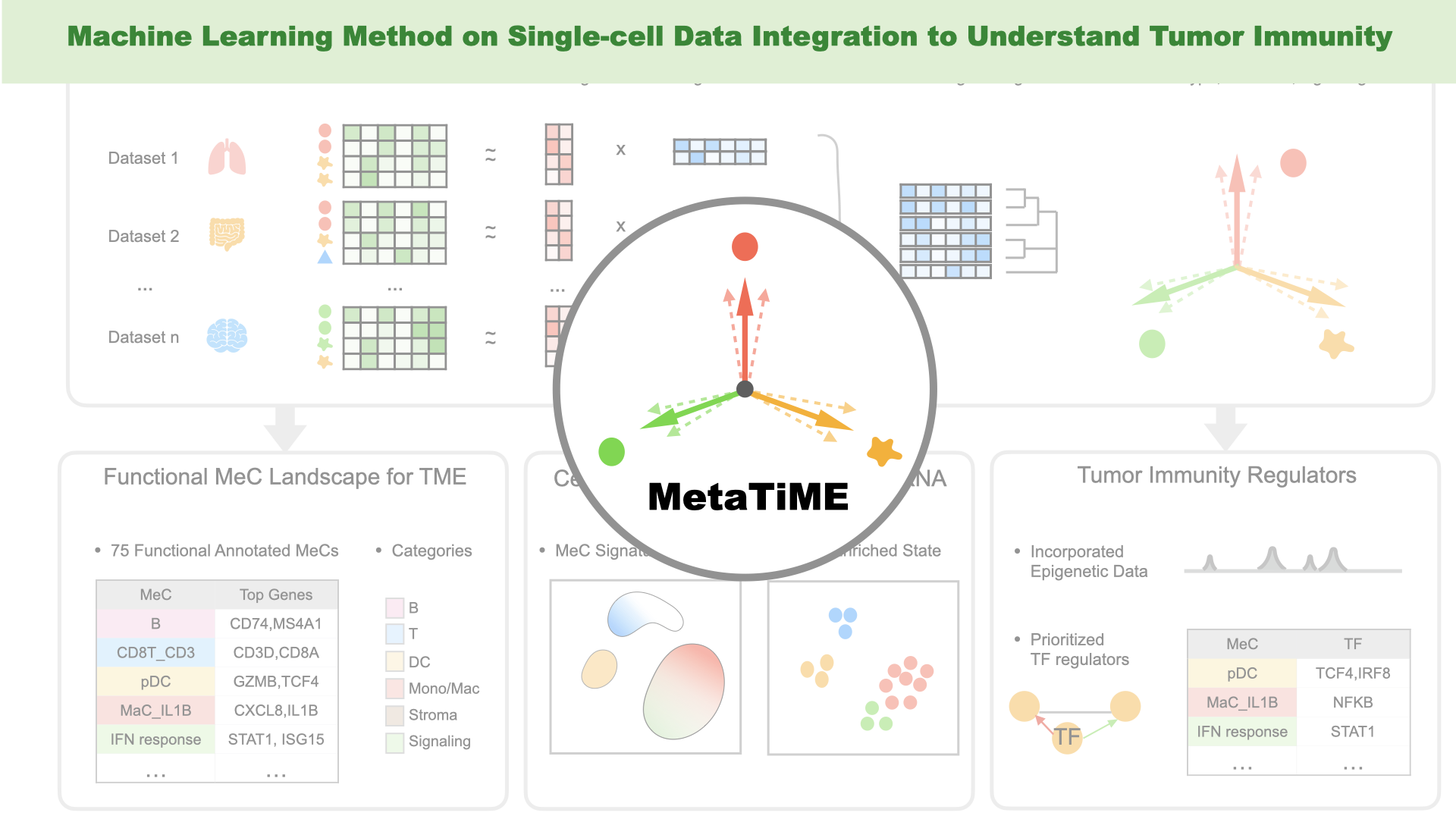
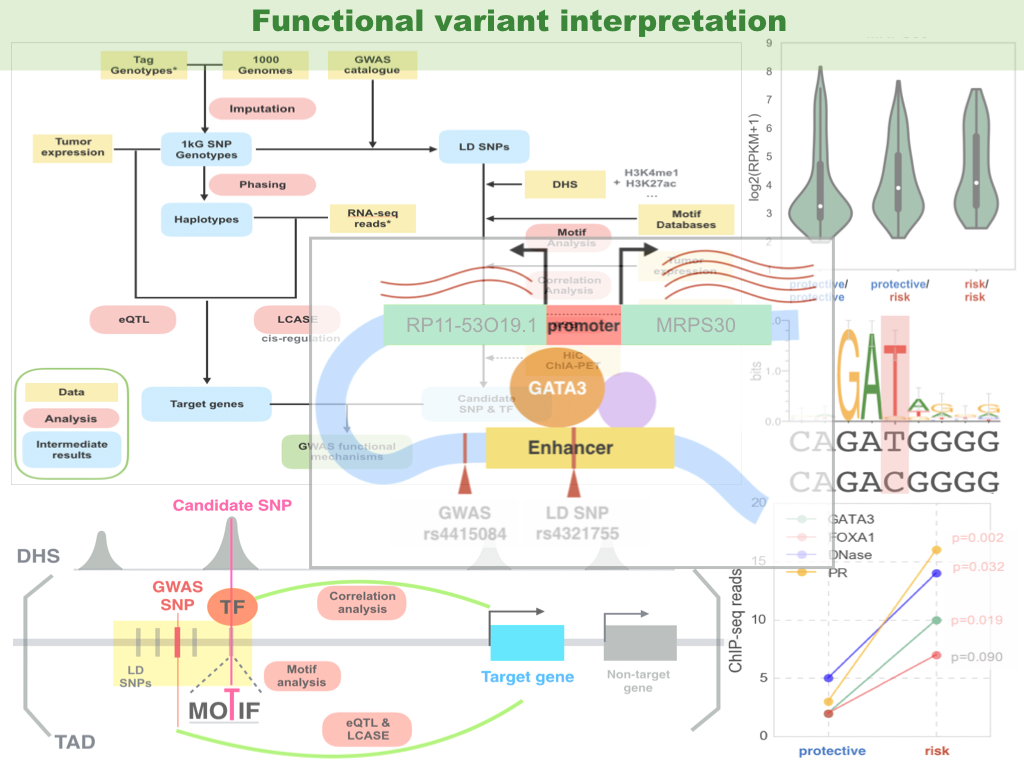
Previous genome-wide association studies (GWAS) have identified common genetic variants that modulate cancer susceptibility, with their causative mechanisms missing. We have developed computational methods for functional interpretation of variants associated with cancer, by combining analyses of expression quantitative trait loci (eQTL), a modified version of allele-specific expression (ASE) that systematically utilizes haplotype information, transcription factor (TF) binding preference, and epigenetic information. Databases/techniques useful in this study are: TCGA, ENCODE, GTEx, Roadmap genomics, TF motif databases, genotyping array, RNA-seq, ChIP-seq, ChIA-PET, Hi-C, etc.. Our computational framework provides an effective means to integrate GWAS results with high-throughput genomic and epigenomic data.
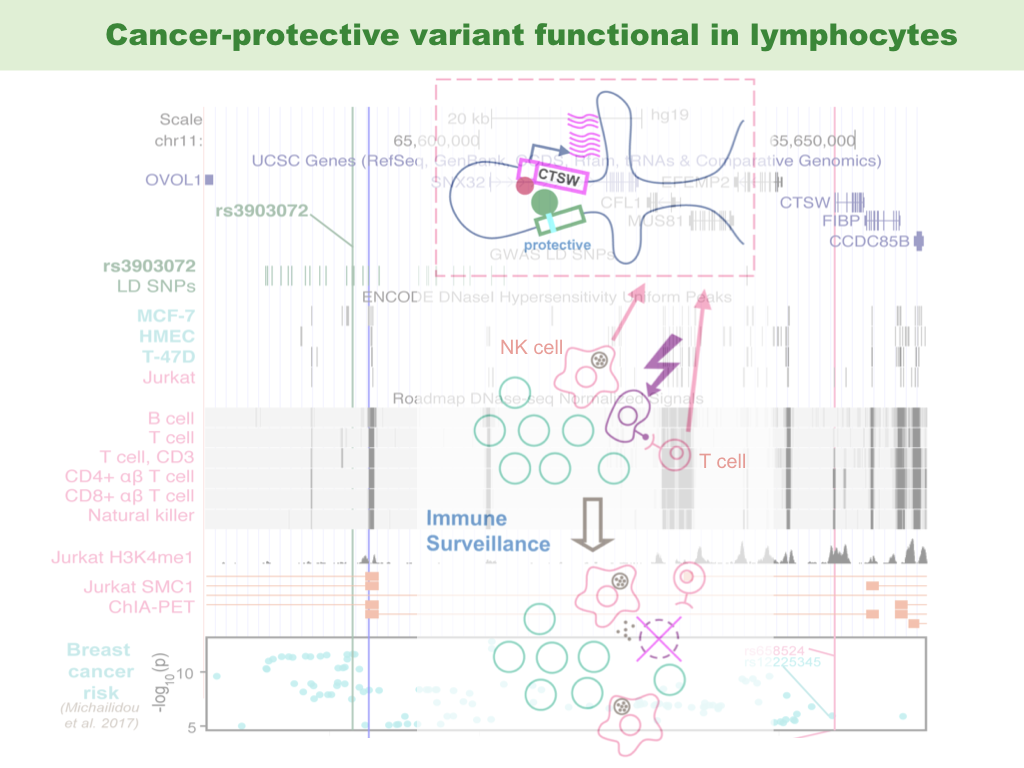
Functional interpretation of non-coding cancer variants has been successful, thanks to the epigenetic information in corresponding cancer cell types and matched normal tissues. However, this approach does not explore the potential effect of risk germline variants on other important cell types that constitute the microenvironment of tumor or its precursor. We show evidence that a breast cancer-associated variant may regulate a tumor-suppressing gene in tumor infiltrating lymphocytes, in particular, T lymphocytes and natural killer (NK) cells. Our hypothesis poses the possibility that cancer variants could be functional in immune cells in the tumor microenvironment, thereby modulating the immune surveillance and affecting the clearing of early cancer initiating cells. This was an idea flashed into my mind after a journal club in the Song Group. Thanks the immune cells for surveillance.
[ Zhang et al. Bioinformatics 2019 ] [ SequencEnG page ]
Next-generation sequencing (NGS) techniques are revolutionizing biomedical research by providing powerful methods for generating genomic and epigenomic profiles. However, a neat introductory learning resource is lacking. We have developed an interactive online educational resource called SequencEnG (acronym for Sequencing Techniques Engine for Genomics) to provide a tree-structured knowledge base of 71 different sequencing techniques and step-by-step NGS data analysis pipelines comparing popular tools. SequencEnG is part of the project KnowEnG (Knowledge Engine for Genomics). I wish I could have a resource like this when I first entered bioinformatics :D
Update 2022-10: SequencEnG has 10,985 users Worldwide!
Related Courses
| Mathematical Statistics | Machine Learning | Computational Cancer Biology |
| Stochastic Processes | Statistical Learning | Statistical Data Analysis in Physics |
Course Projects
Statistical Learning: Prediction of Movie votes and campus shooting with SVM and gradient boosting models.Machine Learning: Classification of NGS technique articles using natural language processing and SVM models.
Stochastic Processes: Segmentation of copy number using HMM based on genotype data in cancer cells.
Statistical Data Analysis in Physics: A cross-entropy method for graph clustering and Cheeger constant estimation.
Workshop
NHGRI Short Course on NGS: Technology & Statistical Methods, Birmingham, AL, 2016.New England Future Faculty Workshop, Northeastern University, Boston, MA , 2020.
Teaching
- Teaching Assistant, course materials Harvard, High Impact Cancer Research Course, 2020
- Invited Speaker, Bioinformatics Seminar, UIUC ANSC590, 2016, 2018
- Speaker, Bioengineering Graduate Student Seminar, UIUC, 2018
- Teaching Assistant, Biomedical Instrumentation Lab, UIUC BIOE415, 2014-2015
Honors and Awards
- EECS Rising Star Awardee, EECS Rising Star Workshop , University of Texas at Austin, 2022
- Emerging Scholars in Genome Sciences Symposium Center for Public Health Genomics, University of Virginia, 2022
- Best Poster Award, Bioengineering Graduate Annual Symposium, UIUC, 2017
- UCLA-CSST Summer Research Scholarship, UCLA, 2013
- Outstanding Undergraduate Student Honor, USTC, 2013
- Outstanding Student Scholarship, School of Life Sciences, USTC, 2013, 2014
- Aegon-Industrial Foundation Scholarship, USTC, 2012
- Panasonic Elite Scholarship, USTC, 2011